Size Exclusion Chromatography (aka Gel Filtration): theory & practice of preparative & analytical

Vložit
- čas přidán 16. 03. 2022
- Of all the types of protein chromatography you may be usin’, size exclusion tends to cause the most confusion! Because, unlike in gel electrophoresis (e.g. SDS-PAGE), it’s the bigger proteins that win the gel mesh drag race when it comes to the size exclusion case! And, no matter what type of SEC (aka gel filtration) you’re doing (i.e. preparative or analytical), choosing the right column is critical!
blog form (with full text & figures): bit.ly/sizeexclusionchromatog...
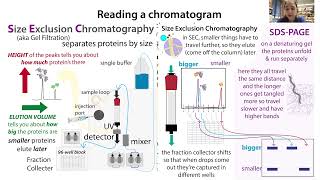
SEC is a column-based method used to separate proteins by size/shape - either for:
1. purification (preparative SEC), often as a “polishing step” after you’ve done other separations (e.g. affinity and/or ion exchange) or
2. to see if proteins interact (analytical SEC)
Unlike with affinity chromatography and ion exchange chromatography, where the proteins actually bind (reversibly) to the resin (either because of some unique feature like an affinity tag (e.g. 6XHis or Strep) in the case of “affinity chromatography,” or because of charge in the case of ion exchange, with size exclusion chromatography (SEC, aka gel filtration (GF)) the proteins and beads don’t actually directly interact, they just take different routes.
Smaller proteins have to go the long way, so it takes them longer to get through and they elute (come off the column) later - which we see as peaks on the chromatograph. This chromatograph shows light absorption measured as the protein passes through a UV detector after coming off (eluting from) the column en route to a fraction collector.
You can think about it as having lots of cars traveling to the same destination - the fraction collector. The route has tunnels with different clearance heights. Bigger “cars” are too big to fit through the tunnels, so they’re “excluded” from a lot of the roads. Thus they miss out seeing a lot of the inside of boring beads and therefore they have shorter trips and get to the final destination more quickly.
The tunnels in SEC are tunnels through porous beads. The beads are often made up of agarose or some other sugar or some polyacrylamide if you need something stronger and/or finer. From the protein’s perspective, they’re pretty “boring” as is - like well-paved roads they won’t slow the proteins down.
The “rules of the road” in SEC are that cars have to travel the longest route they can - if they can fit through a tunnel they have to go that way. On the bright side, at least those roads are less crowded, right?
But imagine you have car-carrier truck. If it’s empty, it’s not that tall, so it can fit through the tunnels. And if the cars it will carry are on their own, they can fit through no problem as well. But put the cars on the truck and now you have a bigger, taller complex that can’t fit through anymore. So it’s route’s restricted. It can’t go through as many tunnels, so it takes a shorter route and arrives more quickly at the destination. This is the basic of analytical size exclusion chromatography.
That’s what I’m doing today, but usually I’m using preparative SEC - I separate all of my almost-pure protein by size to make it almost perfectly pure and keep all of that precious pure protein. But in analytical SEC, it’s more like PAGE in that you’re using it on just a small bit of sample to “get a look.” SDS-PAGE is a common technique for separating proteins in a gel slab to look (after staining) at the size & number of proteins in the sample. It’s denaturing - the SDS & heat unfold (denature) the proteins - so if you see multiple bands you can’t actually tell if they represent proteins that were actually interacting, or were just both present in the sample. bit.ly/sdspageruler
But with SEC, you don’t unfold the proteins, so protein-protein interactions can hold. The interactions have to be strong enough to withstand the journey. In analogy-terms, the cars on the truck bed have to be at least somewhat held down so they don’t fall off - thankfully in the tunnels there’s not far to go so the cars, if they temporally slip, can get back on fairly easily.
Of course, if 2 proteins elute together it could just mean that they’re similarly-sized. But if they are significantly differently-sized they shouldn’t come out together unless they interact. What you usually do is run a sample of each protein on its own. The smaller protein will elute at a larger volume (later timepoint) & the bigger at a smaller volume (shorter timepoint). When you mix them, the peaks will shift.
A leftward shift in the chromatograph indicates binding - but not “too big” of a shift - if the elution is really early, instead of a nice interaction it indicates aggregation - proteins clumping together so that they can’t go through any tunnels. The void volume, representing what was already in the beads before you started & what went through without going through any tunnels, depends on the column size.
more info: 10.1007/978-1-4939-6412-3_2 ; bit.ly/3udiEZA - Věda a technologie








Thank God I discovered your channel! You are amazing, Brianne! I'm a cellular molecular biology undergrad and was kind of struggling with the whole process of using the chromatography machine. We have NGS one, so I guess it looks a little different from yours but works the same. I haven't watched all of your videos yet, but if you haven't made it already, it would really be great if you could make a video doing a protein purification and explaining the reasoning behind each step as you do it. Thank you again & I'm really inspired by you!
Thanks so much! I'm really happy I could help. I have a page of posts on my blog discussing protein purification you might find helpful: bit.ly/proteinpurificationtech Here's a video on using an AKTA: bit.ly/aktainaction And one on the protein purification workflow: czcams.com/video/U5R7t_AYogw/video.html I also have a playlist on protein purification. Hope something there can help!
performed GFC for the first time this year but we used sephadex G-50 also we don't have an automated column or digital detector so we packed our own column with gel and poured the buffer and eluted the sample in small test tubes and took their absorbance readings one by one on the spectrophotometer (we had around 40 labelled fractions!) we even plotted the peaks on a graph paper and everything .
(idk why am i telling u all of this great vid btw thanks for ur insights)
Oh wow! That's quite the effort you must have put in. Hope it was successful! (and thank you!)
love to watch your videos! It is very educational for me to watch :)
Great to hear - thank you!
Hey, Brianne. Great video. May I know how do you make all these infographs/figures?
Thank you! I use Adobe Illustrator and I have some videos on it.
How many fold does your sample get diluted during a sec run with the sec columns you use most? Also, does the subsequent concentration step lead to just as much or less aggregation of your protein compared to what you were able to separate out during your first sec run? Love you videos! Thanks!
Thanks! Dilution before/through will depend on the sample volume and loop volume and column packing and stuff. Not really sure. Otherwise "dilution" can actually just be proteins separating. I typically inject ~500uL and concentrate ~2.5mL. I don't usually get precipitation during the subsequent concentration. Hope that helps
Really great vlog! Our lab is just thinking of setting up a low-pressure chromatography set-up LC set-up but is sad that the system from bio-rad has been discontinued. Can you recommend a good system for us to consider? I have a beginner's knowledge of the equipment and am scratching my head with lots of options, my goal is to do SEC from my cell culture supernatant and other is to enrich proteins for glycosylation and perform purification.
Thank you so much! Maybe the AKTA Start system?
Hi, Thank you for the video. I have a question, I am having problems with the purification, I am purifying a protein doing IMAC and then Amilose, and at the end SEC, my PI suggested that I may not inject the sample at the right time; for SEC, what is the optimal time to inject the sample?
You can inject it into the loop "whenever", but wait until the column is fully equilibrated before putting it on the column
Great video..
Could you please male video on nucleic acid Binding protein purification.
How do you manage to get it RNA free. As I see it on Twitter.
Thanks! It depends on the protein. In my case the protein binds very strongly to endogenous RNAs, so I separate the RNA-bound from the endogenously-free with cation exchange chromatography. If the binding is weaker, you can try using high salt to get the RNA un-stuck
size exclusion chromatography using a syringe rather than FPLC .. would you happen to know how that works ?
The syringe is just to inject it into the FPLC